Low or no data? Visit zero.govt.nz, scroll down the page then click on our logo to return to our site and browse for free.
Cystic fibrosis
Also known as mucoviscidosis
Key points about cystic fibrosis (CF)
- Cystic fibrosis (CF) is an inherited disorder that creates breathing and digestive problems by clogging organs in the body with a thick, sticky mucus.
- CF is the most common life-threatening inherited disorder in New Zealand. Approximately 1 in 25 people carry the CF gene.
- It does not affect everyone equally and symptoms vary from mild to severe.
- CF is a long-term condition that has a huge impact on a person’s life. However, with appropriate treatment, care and support, most people with CF still manage to live a normal life.

Cystic fibrosis is caused by a problem or mutation in one of our genes.
- The specific mutation for CF is found on chromosome 7 and is known as the ‘cystic fibrosis transmembrane conductance regulator’ (CFTR) gene. The CFTR gene helps move salt in and out of your cells. In CF, the movement of salt doesn’t happen properly.
- At least 1,700 mutations of the CFTR gene have been found.
- Different mutations can influence how you are affected, but knowing your mutation can’t predict how severe your symptoms will be.
One in 25 people in New Zealand carry the CF gene. Often people do not know they carry the CF gene, as it causes no symptoms.
To be born with CF, a child needs to inherit two faulty genes: one from their mother and one from their father.
If both parents carry the CFTR gene, with each pregnancy there is a:
- 1 in 4 chance that your child will have CF
- 2 in 4 chance that your child will be a carrier of the CF gene
- 1 in 4 chance that your child will not have CF and will not be a carrier.
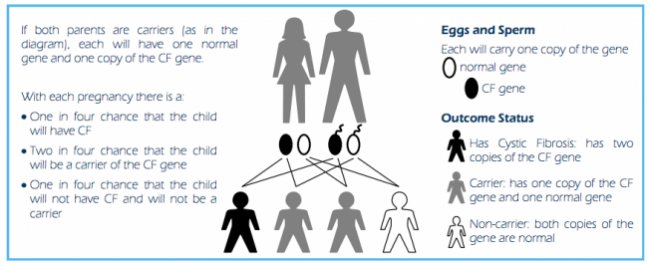
Image source: Genetics of cystic fibrosis(external link) a layperson’s guide Cystic Fibrosis New Zealand.
CF affects 1 in 3,500 newborns. Among different ethnic groups:
- CF is the most common inherited disease in European or Caucasian populations.
- CF is less common in African, Maori and Pacific populations.
Symptoms of cystic fibrosis vary and depend upon age and disease severity.
In babies, symptoms can include:
- delayed growth or failure to gain weight normally
- no bowel movements in first 24 to 48 hours of life
- salty-tasting skin.
In children and adults symptoms may include:
- weight loss or poor weight gain or growth
- severe constipation and abdominal pain
- nausea and loss of appetite
- increased gas or bloating or a belly that appears swollen (distended)
- stools (poo) that are pale or clay coloured, foul smelling, have mucus or that float
- infertility (in men)
- fatigue
- coughing and increased mucus in the chest
- recurrent chest infections or pneumonia
- sinus pain and infections (50% of people with CF have nasal polyps).
Complications from cystic fibrosis often affect the lungs and digestive system.
In the lungs CF can cause:
- chronic cough
- shortness of breath
- repeated chest infections
- bronchiectasis.
In the digestive system CF can block or reduce the release of enzymes from the pancreas that helps to break down food. This is called pancreatic insufficiency, which affects 85% of people with CF. This leads to poor weight gain and malnutrition.
Other complications include:
- nasal polyps, which affect 50% of people with CF
- liver disease, which usually affects up to 30% of people with CF by adulthood
- CF-related diabetes, which is usually present in 2% of children, 19% of adolescents and 40-50% of adults with CF
- progressive respiratory failure.
The diagnosis of cystic fibrosis is made through a range of tests.
Guthrie heel prick
Most babies with CF are diagnosed through a screening test done shortly after birth. This test, called the Guthrie heel prick, checks for many genetic disorders. If a heel prick test is abnormal your child will be referred for further tests.
Sweat test
The sweat test is another way of making a diagnosis of cystic fibrosis. Here, an abnormally high level of salt is found in a child or adult's sweat.
Chorionic villus sampling
In women who are pregnant, a special test known as chorionic villus sampling may be performed. A small amount of fluid is removed from the womb and is analysed for the presence of the CFTR gene.
Genetic testing
Genetic testing through a blood test can also be offered to family members, potential partners or to help detect carriers of the CFTR gene. Genetic testing is also available to people without a family history of CF. It is not publicly funded and the finding should be used together with genetic counselling.
X-ray and sputum analysis
Patients who develop chest infections may require an x-ray and analysis of the sputum to detect the presence of any bacteria.
Cystic fibrosis is a life-long condition for which there is currently no cure. Treatments and life expectancy have dramatically improved over recent years. New drugs are being researched and developed that specifically target the genetic mutation, meaning they treat the actual cause of CF and not just the symptoms.
The goals of CF treatment include:
- preventing and controlling lung infections
- clearing the thick, sticky mucus from the lungs
- reducing inflammation and improving lung function
- preventing or treating blockages in the intestines
- ensuring enough nutrition and preventing dehydration (a lack of fluid in the body).
Treatment options for CF include:
- antibiotics
- inhaled enzymes (called Pulmozyme)
- insulin to manage diabetes
- inhalers to open up the airways
- steroids to help reduce swelling within the airways
- physiotherapy and postural drainage of the chest.
- diet tailored to your needs – often a special diet that is high protein and calories is needed
- pancreatic enzymes to help absorb fats and protein
- vitamin supplements, especially vitamins A, D, E and K
- nasal steroids for, or surgical removal of, nasal polyps.
Treatment can be complex and is best is coordinated by a specialist team working alongside the patient, their family and their doctor.
Cystic fibrosis – Jonny's tips
Jonny has four key tips that help him keep well so he is able to enjoy his passions of sports, online personal training and body building.
(Health Navigator Charitable Trust, 2014)
Unfortunately, cystic fibrosis cannot be prevented. If the parents are both carriers of the gene, then there is a 1 in 4 chance that the child will get the condition. However, steps can be taken to ensure that the child leads a healthy and normal life. This can include:
- chest physiotherapy and postural drainage of the lungs
- constant cycles of antibiotics to help prevent infections from occurring
- immunisations to help reduce the chance of developing new infections
- maintaining good nutrition and an ideal body weight to help fight off infections.
Stories from people living with cystic fibrosis.
Video: Young Bruno living with cystic fibrosis
This video may take a few moments to load.
(Tangata Pasifika, NZ, 2019)
Video: Living With Cystic Fibrosis (Being Me: OJ)
This video may take a few moments to load.
(Attitude, NZ, 2021)
Nicole's story – even cystic fibrosis can't stop her passion for helping animals
Nicole was a Respiratory Achievers Award winners back in 2014, and has not let cystic fibrosis stop her passion for helping animals. She tells her story:
"I was diagnosed with the disease cystic fibrosis when I was 3 days old. This chronic disease affects many organs of the body but predominantly causes mucus to build up in the airways, causing frequent infections of the lungs and sinuses. I also have asthma.
I manage my condition by doing daily chest physiotherapy, sinus washouts, nebulising antibiotics, taking a cocktail of pills and inhalers and am admitted to hospital for intravenous antibiotics usually twice a year. I know the importance of exercise and keep fit by mountain biking, playing soccer, netball and running my 2 dogs every day.
I began volunteering at a vet clinic when I was 12 and by 16 was studying veterinary nursing. I volunteered as the chairperson and secretary of Cystic Fibrosis NZ for 5 years helping others with cystic fibrosis in the Wellington region.
My passion is animals and I am heavily involved as a volunteer at the Wellington SPCA. I foster sick kittens and other animals from the SPCA at my home and nurse them back to health so they can have a second chance at life. I am an animal therapist and take an SPCA puppy or kitten to visit the elderly at Te Hopai rest home every week, and also volunteer as a vet nurse and am currently in the mobile sterilisation clinic in Lower Hutt helping sterilise animals from low-income families.
I am an emergency reservist and have completed training so I can be deployed in a natural disaster to save animals. I am also a volunteer emergency night responder for the SPCA and 1 night a week (6 pm till 8 am) I am on call responding to animal emergencies. I also work part time as a vet nurse at a private clinic and as an administrator to pay the bills!
I received a scholarship to study a Diploma in Business (which I am currently studying correspondence through SIT). Most days there just aren’t enough hours in the day to fit everything in.
In 2015 I pushed the boundaries of my condition and spent 4 months volunteering in South America as a vet nurse helping raise awareness against animal abuse, treating street animals and visiting rubbish tips to help the abandoned dogs.
I knew it was a real risk coming to a third world country when I have cystic fibrosis, but this was something I had to do … helping animals is just in my blood."
Nikki's story – How a new pair of lungs changed her life
In 2016, Nikki and Kristie, who are known as the ‘cystic sisters’, created a CF funding and awareness campaign. Their ‘We are two sisters doing 65 days of good deeds for cystic fibrosis’ started as a Facebook page, but became much more.
The sisters went around the Tauranga community doing good deeds, such as paying for people’s parking and toll gate fees, and leaving vouchers on cars outside the supermarket. They also spread their goodwill by baking for people, and collected and delivered fruit to organisations like the homeless shelter.
At the time of the campaign, Nikki was waiting for a new lung transplant: her lung function was below 30 per cent of full capacity. She had limited mobility because of her need for oxygen, but made the most of the time she could spend away from being hooked up to respiratory equipment.
The campaign included staging a bubble day at the park where they made bucket-loads of bubble mixture and handed wands and cake pops to children at the event.
At the end of the bubble day, Nikki nearly ran out of energy as she’d had more than enough time away from her oxygen. But it was worth it, she says, to build awareness for CF and raise funds to research for better treatments and ultimately, a cure.
The pair raised more than $1000 for Cystic Fibrosis Tauranga.
“65 days of making lots of people smile was an amazing feeling,” Kristie says.
Nikki had been on a waiting list for a lung transplant for a year-and-a-half when she got the call she’d been waiting for.
“When we got the call we freaked out! We were shaky and panicked, but within 20 minutes we were on the road,” says Kristie, who is also waiting for a lung transplant.
Information sourced from Asthma & Respiratory Foundation NZ.(external link)
Cystic Fibrosis(external link)(external link) NZ
Cystic Fibrosis(external link) Australia
Cystic Fibrosis(external link)(external link) US
Resources
Genetics of cystic fibrosis(external link)(external link) Cystic Fibrosis New Zealand
A guide for parents and caregivers of children diagnosed with cystic fibrosis(external link)(external link) Cystic Fibrosis, Ireland, 2014
References
- Types of CFTR Mutations(external link)(external link) Cystic Fibrosis, US
- Cystic fibrosis frequently asked questions(external link)(external link) Cystic Fibrosis, NZ
Newborn screening and diagnostic protocol for cystic fibrosis in NZ (pdf)(external link)(external link) National Screening Unit, NZ, 2016
More detail about investigations, diagnosis and treatment options(external link)(external link) Patient Info, UK
Brochures

Cystic Fibrosis New Zealand

A guide for parents and caregivers of children diagnosed with cystic fibrosis
Cystic Fibrosis, Ireland, 2014
Need help now?



Credits: Healthify editorial team. Healthify is brought to you by Health Navigator Charitable Trust.
Last reviewed:
Page last updated:
